Introduction
If you've ever heard of bioinformatics, computational biology, protein structure, and the folding problem, then you know what we're going to talk about here.
If you are interested in deep learning and neural networks, their application for solving urgent practical problems and are deeply convinced that if artificial intelligence does not take over the world, it will definitely surpass humans in its cognitive abilities, then you have definitely heard about this victory over human cognition.
Our article will focus, as the name suggests, about the artificial intelligence AlphaFold and its successor AlphaFold2 from DeepMind, which was created to predict the three-dimensional structure of a protein. In 2020, AlphaFold2 performed as well as anyone before, and beat its rivals with devastating performance in a competition dedicated to this problem. No one else found a better word than “ breakthrough ” to describe the results of AlphaFold2's work. And DeepMind's press release about winning this competition caused heated discussions not only among the professional community and sympathizers, but also among people far from worries about predicting the structure of proteins, biology and everything connected with them.
Here we will discuss what this structure prediction problem is and why it is so difficult. Let us tell you why it is important not only for the academic community, but also for what is commonly called the "industry". And, of course, we will also express our opinion about what this breakthrough in artificial intelligence means for science and industry in general, and for us - ordinary people who sometimes get sick and are treated for their diseases, in particular.
Protein structure: what is it and why is it needed
Since Habr is a resource primarily about IT and everything connected with it, in order to understand all the commotion that AlphaFold2 caused, it makes sense to cover a little the subject structural and biological area.
Let's start with the hardest part - defining a protein and describing its structure. Proteins, they are also polypeptides, they are proteins - molecules that ensure the course of most of the processes in our body. They can transmit and receive signals that the cells of our body exchange, can participate in metabolism (such as insulin , which is responsible for maintaining glucose levels in the body). And even antibodies- those active participants in the immune response to various pathogens are also proteins. In general, proteins are always, proteins are everywhere. But why are they so powerful and versatile?
What a protein does and how well it does it is determined by its composition and structure. There are as many as 4 types of them.
The primary structure is the so-called protein sequence. Proteins are made up of 20 standard building blocks called alpha amino acids . Each of the amino acids is designated in the form of a letter: for example, glycine - G, alanine - A, and asparagine - N. So it turns out that for each protein we can write down a line from the 20-letter alphabet:
Insulin: MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
PD-1: MQIPQAPWPVVWAVLQLGWRPGWFLDSPDRPWNPPTFSPALLVVTEGDNATFTCSFSNTSESFVLNWYRMSPSNQTDKLAAFPEDRSQPGQDCRFRVTQLPNGRDFHMSVVRARRNDSGTYLCGAISLAPKAQIKESLRAELRVTERRAEVPTAHPSPSPRPAGQFQTLVVGVVGGLLGSLVLLVWVLAVICSRAARGTIGARRTGQPLKEDPSAVPVFSVDYGELDFQWREKTPEPPVPCVPEQTEYATIVFPSGMGTSSPARRGSADGPRSAQPLRPEDGHCSWPL
Knowing the DNA sequence (or rather, the RNA can uniquely identify this same sequence of letters for proteins using a table of the genetic code :
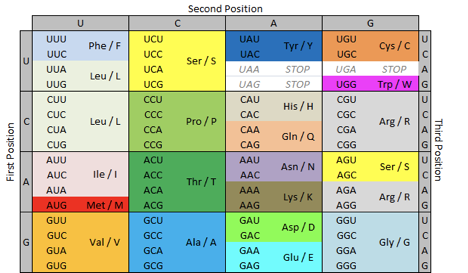
TABLE translation triplets (t) amino acid residue at nucleotides
But letters, and even lines, are not enough for us. To understand how a protein works, with what and how it can interact in the body, we need to know how it works in space.
Each amino acid can be represented as a set of atoms, located in a certain way in such a familiar three-dimensional space. Each atom has its own coordinate and its own set of bonds with neighboring atoms. This is how threonine looks, for example:

The structure of the
amino acid threonine in atomic representation Amino acid sequences are able to fit in space into stable secondary structures - the so-called alpha-helices and beta-layers (and all sorts of different ones), which are usually visualized with graceful ribbons:

Alpha-helix

Beta-fold
A whole protein, consisting of many amino acids, and, therefore, many atoms with their own coordinates, may look like this:

The way a complete amino acid chain is folded in space is called a tertiary structure . Everything is sewn into it: what residues and atoms the protein consists of and how they are located relative to each other in three-dimensional space.
There are even more complex proteins - consisting of several polypeptide chains and capable of functioning only in this form. The structure of such proteins in three-dimensional space is called quaternary . A prominent representative of such proteins is hemoglobin, which carries oxygen through our tissues:

The structure of hemoglobin
It is the complete spatial structure of the protein (tertiary or quaternary) that is commonly called simply structure. And it is she who completely determines its function. From the three-dimensional structure of the protein, it becomes clear which amino acids are capable of forming interactions with other substances, how strong these interactions will be and what these interactions will lead to: for example, to the cleavage of other proteins or maintaining the correct folding of DNA molecules in the cell.
Knowing the structure of a protein, one can rationally approach the creation of a drug that should interact with it. Or tweak a cool existing animal protein so that it doesn't elicit an immune responsein a person and treat a person with it. And if you have the ability to build a protein structure in sequence, you can even make your own artificial protein that will perform a function for which there is no natural protein - for example, to break down plastic. In general, the room for maneuver is unlimited and the horizons are wide.
That is why many scientific (and not only) groups are fighting over the problem of obtaining a protein structure. The traditional approach to determining the structure of a protein is experimental. This can be done, for example, using X-ray structural analysis (XRD), nuclear magnetic resonance (NMR) or cryoelectron microscopy.(Cryo-EM). All of these methods have their pros and cons. They provide fairly reliable information about how a protein is organized in space, how the atoms in its amino acids are located relative to each other - that's all you need. But they also have three common disadvantages: expensive, long and difficult. Sometimes it is simply not possible to obtain a protein sample in the form required for XRD analysis. Deciphering NMR results for proteins longer than 200 amino acids is still not a trivial task. A good cryoelectron microscope costs several million dollars, and there are not so many specialists who know the art of taming it, even all over the world.
All this means that the question "Shouldn't we predict the structure of a protein by its sequence using bioinformatics methods?" has not won over with its novelty for a long time - there have been a lot of attempts to solve this problem (and, we are sure, there will be many more!). They cannot be called truly successful until 2020, and why this is so, we will talk closer to the end of the article. But the history of what approaches were used and how efficient they were cannot be separated from the history of the competition called CASP.
CASP
Critical Assessment of protein Structure Prediction - or simply CASP - is a competition to predict the three-dimensional structure of proteins from their sequence. It takes place every two years, and all self-respecting groups participate in it that have developed a new algorithm to solve this problem. This event has been held since 1994, then 35 groups took part in it. But in 2020 there are already more than a hundred. What are they competing in?
The organizers invite participants to predict the structure of proteins, knowing only their sequence. These structures have already been obtained by the experimental methods listed above, but so far no one has seen them. Usually, all allowed structures are published in the Protein Data Bank database. , and anyone can find them there. But some experimenters save their results for just such a case.
By participating in CASP, one can show a class in different categories of structure prediction - for example, outperforming everyone in predicting the packing of disordered structures or contacts between different regions of a protein. Or try to predict the tertiary structure of a protein by homology. For example, you can find a protein that is most similar in sequence to the one whose structure you want to predict, and the coordinates of the atoms for which are already known. And then with all sorts of modifications to bring this "template" to the beautiful desired tertiary structure of the required protein. How the template is chosen and what tricks are used in this category, you can find out here: Homologous Protein Folding | Pavel Yakovlev (BIOCAD) .

But the de novo (or ab initio ) prediction of the tertiary structure of a protein is traditionally considered the most prestigious and exciting category for the public . These beautiful Latin words mean that when predicting you do not use directly known structures of proteins, but use certain patterns and rules to build a structure from scratch.
At the same time, the organizers are not very concerned about the complete structure of proteins with the locations of all atoms. They are only interested in the so-called Cα atoms - those from which amino acid radicals grow (in this figure, they are marked in blue):

Amino acid trimer. Cα atoms are marked in blue (hydrogen atoms are removed for the sake of clarity of the picture)
“How so ?!”, you ask indignantly. We read here for 20 minutes about atoms, about coordinates, about how important this is for the function of a protein, etc. and so on, and now let's throw everything out and we will only predict this incoherent set of points!
Now it will become clear why this is possible. Let's first remove these same radicals and see how a protein looks without them:

Polypeptide chain without amino acid radical. Cα atoms are blue (hydrogen atoms removed for the sake of clarity)
In general, the general form of the protein is clear. To restore the direction of what grows from Cα, one can rely on the known structures from PDB (there are more than 170 thousand of them): simply take and, according to the general structure of this protein, choose more suitable radical orientations for it.
And if we remove everything except Cα, it will look like this:

Arrangement of Cα atoms in the protein
Looks bad, but everything is not so hopeless. We know that there are always carbon and nitrogen atoms between two Cα. And thanks to quantum mechanics, we know what the distances of the connections between them are - they are always the same. Moreover, since 1963 [ 10.1016 / S0022-2836 (63) 80023-6 ] we even know what are the permissible angles between the planes (they are dihedral angles) that they form:

Determination of phi and psi angles in a protein

Ramachandran's map - distribution of the observed phi and psi values in proteins as a whole.
So knowing the distances and angles between the missing atoms, it is a matter of technique to reconstruct their coordinates.
So, the CASP participants predict the position of Cα atoms in space from the available amino acid sequence of the protein. Now it's time to evaluate who is the best. There is a special metric for this - GDT_TS.
At the end of the competition, the organizers have real Cα positions from the experiment, and those predicted by the participants. To assess the quality of predictions, you should first combine them with each other, for example, as follows:

Two superimposed structures of one protein
This is done using structural alignment algorithms (for example, this one ). Now you can evaluate the similarity of the resulting structures just by this metric. GDT (X) - Global Distance Test from X is the fraction of those Cα that, after structural alignment, are no more than a given distance X from the reference. In the figure above, for example, one of the predicted Cα is at a distance of 7.3 Å from the reference one. The share, as usual, is distributed from 0 to 100%. GDT_TS - Global Distance Test Total Score in CASP is defined like this:
GDT_TS = 1/4 (GDT (1 Å) + GDT (2 Å) + GDT (4 Å) + GDT (8 Å))
But that's not all. The GDT_TS indicators obtained by the participants' sweat and blood on all structure predictions are converted to a Z-score... In this case, outliers are thrown out and too low values (with thresholds of -2 or 0) are cut off.
The final metrics for each team (and there are 4 of them!) Are the total Z-score for threshold -2, total Z-score for threshold 0, average Z-score for threshold -2 and average Z-score for threshold 0.
It is said that this allows you to become winners, silver medalists, etc. several teams at once. But in 2020, there was only one winner in all respects - AlphaFold2. The number 2 at the end hints that this instrument has a history. Let's talk about it now.
CASP13, CASP14 and DeepMind
In 2018, the CASP13 competition took place, in which, albeit not a devastating, but very convincing victory was won by a team called A7D:
Z-scores of the teams participating in CASP13
This A7D was ... no, not Albert Einstein. And the team from DeepMind , who developed the AlphaFold algorithm. DeepMind is known to anyone interested in artificial intelligence. They were founded only 10 years ago, and in 2014 they were acquired by Google. For the short 10 years of their existence, these guys managed to be noted in many areas. Artificial intelligence AlphaGo won in 2016 world champion in the game of go. This game requires a developed strategic and tactical thinking, previously inherent only in humans. In 2019, after a series of successes in creating artificial intelligence capable of playing video games as well as humans, the AlphaStar neural network became the Grandmaster of Starcraft II in 44 days .
In 2020, on CASP14, the results looked like this:

Nobody saw such numbers on CASP. The indicator looks even more impressive, which does not depend on the results of other teams: the median for all GDT_TS structures for AlphaFold2 was 92.4. And this means (at least in terms ofDeepMind itself) that their prediction accuracy is comparable to the structure resolution accuracy that experimental methods give. But best of all, the quality of structure prediction is reflected, of course, by beautiful and really impressive pictures:

Source
Undoubtedly, the results are cool. Let's see what is hidden under the hood of AlphaFold and AlphaFold2, which allows them to solve this problem with such amazing accuracy.
Alphafold
It is clear that the results of AlphaFold2 are much more impressive, and it would be interesting to understand how exactly it works. But, alas, so far we do not have any detailed information from its creators - only a press release dedicated to the victory in CASP14. However, hints on how it works can be found in the article on the first version of AlphaFold.
Although the victory at CASP13 took place in 2018, an article in the prestigious magazine "Nature", which describes the algorithm of the AlphaFold, came out only in January 2020. So the publication about AlphaFold2 will have to wait a little longer. For now, let's talk about the first version.
AlphaFold is an ultra-precise neural network. Accordingly, it has features that are accepted as input, the architecture through which these features pass, and the final predicted data that comes out of this neural network. Let's start with the input features.
AlphaFold Login
Recall that the task is to predict the structure of a protein from its amino acid sequence. But AlphaFold is not so simple: the neural network accepts not only the sequence itself, but its multiple alignment on sequences from various large databases of existing proteins.
And behind this is a very beautiful idea that the DeepMind team used. It perfectly demonstrates that in order to solve problems using bioinformatics methods, one must not only be a good mathematician and programmer, but also understand their biological foundation.
So the idea is as follows. We take a protein sequence and align it with all protein sequences known to mankind. There are very, very many of them, much more than structures, because, as we remember, it is very simple to obtain a protein sequence, knowing the DNA sequence. And sequencing projects - determining the sequence of DNA for different animals - have generated so much data in recent years that it would be a crime not to use it.
And so, we found proteins similar to each other in sequence. If they are similar in sequence, then they are most likely similar in function and structure. But there is not necessarily a structure for these similar proteins (most likely, there is none). And we don't need it now, because we can deduce other patterns of similarities.
Amino acids of a protein interact with each other - they form stable (and not very) bonds that form, maintain and stabilize its structure.

And if one of the interacting amino acids suddenly changes, moves or disappears, then its counterpart can be upset, displaced and cease to support the structure. This will unravel the protein and lose its ability to function. And if he loses the ability to function normally, the health and quality of life of the organism in which he lives may suffer greatly. And such an organism is unlikely to live and multiply for a long time, and we are unlikely to have time to sequence it.
What conclusion can be drawn from this? And such that if pairs of amino acids form critical interactions, then in similar proteins they either do not change, or change synchronously. Therefore, looking at the alignment of similar proteins, one can calculate the correlation of each pair of positions and build what is called a coevolution matrix. Where the correlation is higher, there are likely to be critical interactions, and therefore these positions are most likely to be in space next to each other.

Illustration of the idea of using information about coevolution to predict the structure Source
The idea, although it does not belong to DeepMind , has shown itself in all its glory in their hands.
AlphaFold exit
Now let's talk about what AlphaFold produces at the output. Let's start, as usual, from afar.
The task requires to predict the position of all Cα proteins. To solve this problem, you can do the prediction of the coordinates of the atoms. But imagine a protein with only one atomic coordinates. Now take it two meters up, one meter to the left, and 78 centimeters forward. And rotate around the center of mass 15 degrees. The protein and structure remained the same, but the coordinates changed a lot. Therefore, predicting three-dimensional coordinates is a thankless task, and no one does this. So we have to come up with something else. And before describing the “other”, let us again plunge into the biological component of the problem and find out what Cβ atoms are.
Cβ - atoms bound to Cα (marked in pink). They have 19 amino acids out of 20. Glycine is content only with Cα.

The good thing about Cβ is that by its distance to Cβ of another amino acid in the protein, one can understand whether these residues interact with each other. If the distance is less than 8 Å - there is interaction, more or equal - no. The opposite is also true: if we know that there is interaction between amino acid residues, then the distance between their Cβ should be less than 8 Å in the final structure. Do you see where we are heading?
The AlphaFold team came up with the idea of predicting pairwise distances between Cβ atoms and at the output the neural network outputs discrete probability distributions of the probability of pairwise distances between these atoms of different residues. For the remainder at number 29, it looks like this:

Source
The distance probability distribution for Cβ is very cool, of course, but we need a structure here, or at least the position of Cα. Not a problem - everything will be done now.
From these probabilities for each amino acid, a distogram is constructed - a matrix of predicted pairwise distances between Cβ:

Source
We know the distances between the remnants, and the structure can be restored. To do this, DeepMind constructed the following potential, depending on the phi and psi angles - those same dihedral angles, the distribution of whose values we talked about earlier:

The first contribution is the potential from the predicted Cb - G (phi, psi) position. This is a function that expresses their pairwise distance in terms of these angles. The second contribution came to us from a small surprise that can only be revealed if you read the article very carefully. Together with pairwise distances, DeepMind predicts the probability distribution of the values of the dihedral angles phi and psi - hence the contribution to the potential. And the third contribution is intended to avoid the situation when all the constructed atoms collide with each other and their coordinates overlap.
This potential is nothing more than a mathematical model describing the potential energy of this protein. There are many such models ( hereyou can see what they are), but the AlphaFold team created their own. The laws of thermodynamics tell us that a closed system tends to a minimum of potential energy. And if we know the functional form of this energy, and, moreover, it is also differentiable, as in this case, then we can minimize it and come to a real structure. So at this stage, a completely dry mathematical problem of optimizing the functional arises. To solve this, the team uses the gradient descent method .
And as a result, there are values of all phi and psi from the optimal structure. We know the distance between Cα, C and N. It is not difficult to reconstruct the correct relative positions of Cα from this.
So, everything is clear with the input features. So does the post-processing of the algorithm results. And what sits inside this neural network, which so accurately predicts pairwise distances Cβ and the distribution of dihedral angles?
AlphaFold architecture
The article provides a diagram of a neural network:

This is a convolutional neural network, which belongs to the ResNet class , which allows it to be trained more efficiently . The training sample consisted of ~ 30,000 experimentally obtained structures from the PDB database, which we talked about above, and the training took about 5 days.
Generally speaking, there was no breakthrough in the construction of the network architecture here - just a powerful and logically built neural network. And its success lies, of course, precisely in the choice of input and output data.
AlphaFold2
And what about AlphaFold2? We do not know much about his device. Schematically, its architecture looks like this:

From the press release, we know that this neural network is 'end-to-end', that is, (probably) it only needs to have a protein sequence as input, on the basis of which it will independently make multiple alignment and pull from it the necessary features. And at the exit, the structure will immediately wait for us.
This time the training took place on approximately 170,000 PDB structures and took several weeks.
And it seems that the idea is still the same - from the data on the correlation of positions, to obtain the probability distribution of pairwise distances. Or not. In general, we will look forward to an article in a prestigious magazine.
Discussion
Unlike parliament, a post on Habré and comments to it are quite a comfortable place for discussion, so here we will express several opinions about all of the above facts. And I want to start by examining what caused the general outrage among those involved in structural and computational biology.
The DeppMind announcement that we've referred so often here is called "AlphaFold: a solution to a 50-year-old grand challenge in biology." And in the very first paragraph it says that this team managed to solve the folding problem. Reading this, many exploded, and for various reasons.
First, strictly speaking, they didn’t solve any folding problem, they didn’t solve it. Folding is a processthe protein acquires its tertiary structure. That is, solving the folding problem is identifying the patterns that lead the protein sequence to various stages of the formation of its structure and to the final tertiary structure. And this task is much more difficult, because proteins sew one amino acid to themselves and begin to form their structure as they grow in a complex multicomponent environment. And they can do this in different ways, depending on external conditions. And they have an infinite number of ways to arrive at the final structure.
At the same time, it is known that proteins do not simply enumerate all possible conformations. Levinthal's paradoxjust says that if a protein of 100 amino acids did exactly this, then even with a wild rate of iteration of conformations, the lifetime of the Universe would not be enough for it to acquire its structure. The main theory, which now prevails in the fields of solving the folding problem, is called the Anfinsen dogma and says that the protein consistently goes to the kinetically achievable energy minimum.
Obviously, the task of predicting the process of structure formation is much more difficult than what DeepMind did, because they were solving the problem of predicting structure. And such a substitution of concepts has caused in some way fair indignation of citizens sympathetic to the process.
For the sake of fairness, a couple of points should be noted. First, the term folding is very often used by computational biologists precisely in the sense of predicting structure . True, this is done in narrow circles, and in a decent society such things are not said aloud. So it can be assumed that the DeepMind press release simply contained inappropriate jargon. Secondly, if you read this release a little further than the first paragraph, its authors just explain that they were solving the problem of predicting the structure, without any processes there. Well, this is all demagoguery and a struggle for the purity of the language. Let's move on to the second and some reflections on the case.
Namely, thinking about the word "decision". When it comes to using neural networks to determine faces or count the number of cats in a picture, we can always understand whether the neural network was mistaken or not. Simply because most people are able to determine from a picture how many cats are on it and whether it is the person they see in front of them. But when we use a neural network to solve a problem for which we do not know the answer, questions arise. How do we understand that the neural network worked correctly and did not break? With what degree of confidence can we use the results of this neural network in our practice? For example, in a drug development project that costs millions (sometimes even dollars) to fail? So far we do not have an answer to this question, therefore, it is probably too early to talk about a solution.
Of course, in both the first and second versions of AlphaFold, a speed is mentioned, designed to distinguish between good and bad algorithm performance. But, unfortunately, there is no data on how it correlates with reality in the public domain. So, alas, there is no talk about the practical and routine use of speech yet.
And yet. It should be noted that this algorithm showed a work that significantly surpasses all previous attempts to solve the problem of predicting protein structure. And this means that the potential of the method underlying this algorithm is enormous, and such a method can be applied not only to this science-intensive and complex problem. This means that with the proper development of such approaches, a very bright and exciting future awaits us!
Conclusion
In conclusion, I would like to say the following - AlphaFold2 is definitely a breakthrough. So far, there are some points that limit its practical application, but it seems that they can be overcome. Of course, for this you need to set such a goal and confidently go towards it. And I want to believe that DeepMind will not abandon its offspring and will continue to develop and improve it. It will be very great if this development proceeds with the same leaps and bounds as before.
Post-conclusion
We hope you were impressed by the existing problems, the approaches to solving them, and the success of these approaches. In our opinion, the most beautiful thing about such events is the demonstration of how correctly applied knowledge from different fields can give incredible results and help in solving the most difficult problems facing humanity.
AlphaFold was born out of the fact that his team members had competencies in biology, physics, mathematics, deep learning algorithms and optimization - that is, in the field of computational biology. Few places in the world are well taught this, and so far nowhere at a sufficient level is taught in Russia. But in 2021, the Higher School of Economics, together with BIOCAD, is launching a master's program in Computational Biology and Bioinformatics., which will teach these disciplines so necessary to meet such ambitious goals.
Students with a strong physical and mathematical background, without chemical and biological training are expected in the magistracy. HSE lecturers will provide the best courses in the country in algorithms, programming, data analysis, and industry staff will tell the special heads of physics, molecular biology, chemistry, as well as special courses on molecular modeling, structural bioinformatics algorithms, systems pharmacology and other important topics for the field. And, which is also important, they will talk about real industrial biological problems and teach you how to use the knowledge and skills gained to solve them.