
A protein from the bacteria Staphylococcus aureus
In late November, Google's DeepMind team announced that its AlphaFold deep learning system had achieved unprecedented levels of accuracy in solving the protein folding problem , a difficult problem in computational biochemistry.
What is the problem and why is it so difficult to solve?
Proteins are long chains of amino acids. Your DNA codes for these sequences, and RNA helps make proteins according to this genetic blueprint. Proteins are synthesized in the form of linear chains, but subsequently fold into complex spherical structures (see the picture at the beginning of the article).
A part of the chain can curl up into a tight spiral, " α-helix . "The other part can bend back and forth to form a wide, flat figure," β-sheet ":
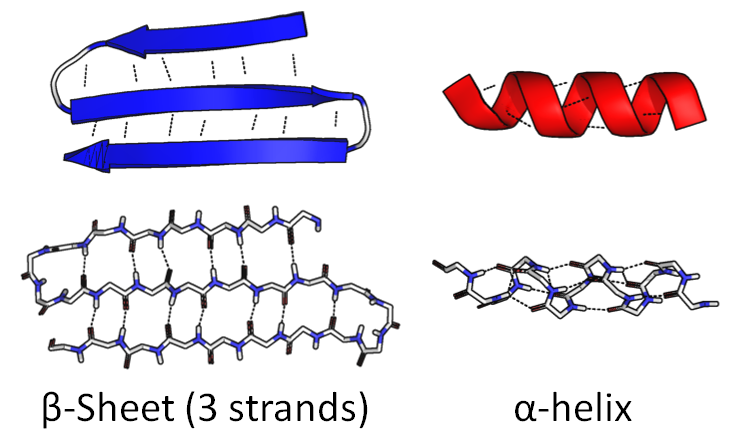
The amino acid sequence itself is called the primary structure . These figures are called secondary structure .
These components themselves also fold to form unique complex shapes. This is called the tertiary structure :
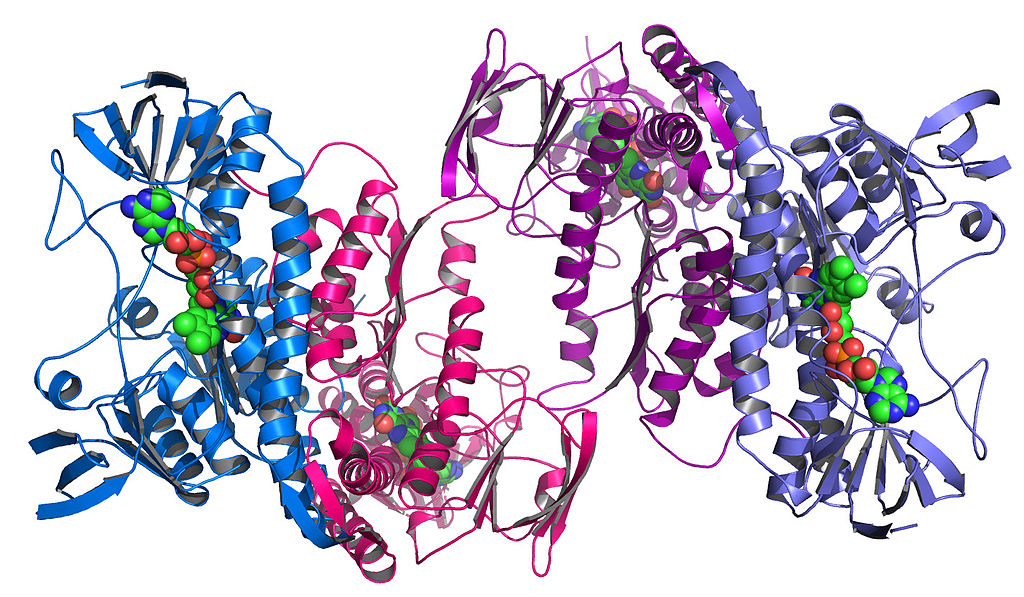
An enzyme taken from the bacteria Colwellia psychrerythraea RRM3

protein
Looks messy. Why is this tangled ball of amino acids so important?
Protein structure is not random! Each protein folds into a distinct, unique, and largely predictable structure, which is essential for it to function properly. Due to its physical form, the protein is well suited to the structures with which it can bind. Other physical properties are also important, especially the distribution of electrical charge over the protein. In the picture, a positive charge is indicated in blue, a negative charge in red:

Surface charge distribution on the lipid carrier protein of plants 1 of rice
If a protein is essentially a self-assembling nanomachine, then the main purpose of an amino acid sequence will be to produce its unique shape, charge distribution, and everything else that determines the function of the protein. How exactly this process takes place is not yet entirely clear - today it is an active area of research.
In any case, understanding the structure is important to understanding how it works. However, the DNA sequence only defines the primary structure of the protein. How do we know its secondary and tertiary structures - that is, the exact shape this tangle will take?
This problem is called the protein folding problem, and there are two basic approaches to it: measurement and prediction.
Experimental methods can measure the structure of a protein. However, this is not so easy to do: structures are not visible through an optical microscope. For a long time, X-ray crystallography was the main method for studying structures. In addition to it, nuclear magnetic resonance was used, and recently a new technology has appeared, cryoelectron microscopy .

X-ray diffraction pattern of SARS protease
However, these methods are expensive, complex and time-consuming, and besides, they do not work with all proteins. In particular, proteins embedded in the cell membrane - the same angiotensin converting enzyme 2 (ACE2) receptor to which the COVID-19 virus binds - folds into the lipid bilayercells, and it is very difficult to crystallize.

The structure of the cell membrane
Therefore, we were able to disassemble the structure of a tiny percentage of the sequenced proteins . The universal protein database contains 180 million sequences, while the database of three-dimensional protein structures contains only 170 thousand positions.
We need a better method.
* * *
Recall that the secondary and tertiary structures of proteins are basically a function of the primary structure that we know from sequencing. What if, instead of measuring the structure of a protein, we could predict it?
This is the task of predicting the structure of proteins. Computational biochemists have been working on it for decades.
How can you approach it?
The obvious way is to simulate the physics of the process directly. We simulate forces for each atom, taking into account its location, charge and chemical bonds. We count accelerations and speeds, and step by step scroll through the evolution of the system. This is called "molecular dynamics".

Supercomputer " Anton " by DE Shaw Research

Supercomputer IBM Blue Gene
Online puzzle Foldit
The problem is that this approach is extremely computationally intensive. A typical protein contains hundreds of amino acids, that is, thousands of atoms. The environment is also important: when folding, the protein interacts with the surrounding water. Therefore, it is necessary to simulate the behavior of about 30 thousand atoms. In this case, an electrostatic interaction occurs between each pair of atoms, that is, with a rough estimate, we get 450 million pairs, a problem with complexity O (N2). There are clever algorithms that reduce its complexity to O (N log N). In addition, 10 9 -10 12 steps must be calculated for the simulation . Exceptional headache.
Okay, but we don't need to simulate the entire folding process. Another approach suggests finding a structure with minimal potential energy. Usually objects tend to come to rest with the least energy, so this heuristic approach is justified. Energy can be calculated by the same molecular dynamics model, which gives us the magnitude of interactions. With this approach, we can try a bunch of candidates and choose the structure with the least energy. The problem, of course, is where to get the structures from. There are simply too many of them - molecular biologist Cyrus Levintol has calculated that there could be about 10,300 . Naturally, you can use a smarter approach than random brute force. But there are still too many of them.
Therefore, many attempts have already been made to speed up such calculations. Anton, a supercomputer from DE Shaw Research, uses special hardware - special integrated circuits. IBM is also using the Blue Gene bio supercomputer. Stanford launched the Folding @ Home project, using the distributed power of home computers. UW's Foldit project turned folding into a game to add human intuition to computation.
Yet, for a long time, no technology has been able to predict a wide range of protein structures with high accuracy. At the CASP competition held twice a year, where the results of the algorithms are compared with the structures measured experimentally, the first places received predictions with an accuracy of 30-40%. Until recently:

Best team median predictive accuracy in the free modeling category.
How does AlphaFold work? It uses multiple deep neural networks to learn different functions associated with each protein. One of the key functions is to predict the resulting distances between pairs of amino acids. This brings the algorithm to the final structure. In one variant of the algorithm (described in the journals Nature and Proteins ), the potential function of this prediction was derived, to which the simplest gradient descent was applied, which worked surprisingly well.
The main advantage of AlphaFold over previous methods is that it does not need to make assumptions about structures. Some methods work by breaking up proteins into sections, counting each one, and then putting everything back together. AlphaFold doesn't need this.
Apparently, DeepMind considers the folding problem to be solved, which seems to me to be an oversimplification, but in any case, their progress is significant. Experts not affiliated with Google use epithets like “ fantastic ” and “ revolutionary ”.
Genetic engineering now has two powerful tools, CRISPR and protein folding. Perhaps the 2020s will be for biotechnology what the 1970s were for computing.
Congratulations to DeepMind researchers on this breakthrough!